parser.h File Reference
Detailed Description
parser commandsAnything in square brackets is optional, and commands separated by slashes (e.g. fc/fs) denote the available options for that command.
Commands can appear in any order in the file, with the exception of the calculation/execution commands, so be sure to give that command last!
Quick reference:
- Input/Output
- Default MTZ labels
- Debugging
- Running the program
- On/off switches
- MTZ file symbols
- Atom selections
- Refinement/parameter setup
- Additional constraints
- Crystallographic settings
- comments: anything following a # symbol is ignored.
- read
- cif lib "identifier1" "identifier2"...
- read in additional cif identifiers (located by searching through the libpath list)
- mtz "file1" "file2"...
- set up a list of MTZ files to read in (the files are read in when needed, so its OK to define the columns to read in at any time).
- if no MTZ files are provided and are required for the requested calculation, then the program will attempt to find any mtz files in the current directory and look for any columns labelled with the default types.
- cif/pdb fc "file1" "file2" [select "groupname"]
- read in a CIF or PDB file for use in structure factor calculations for Fc (fc). If you'd like to only select a subset of the atoms for the calculation, use the optional select tag and define a group to select.
- if no PDB or CIF files are provided and are required for the requested calculation, then the program will load all the PDB files in the current directory.
- cif lib "identifier1" "identifier2"...
- write
- mtz "file"
- output a MTZ file with all the current read in/calculated data to the given filename.
- pdb "file"
- output a PDB file including any shifts (e.g. after refinement)
- cif "file"
- output a CIF file including any shifts
- mtz "file"
- "FO,SIGFO": Fobs
- "I,SIGI": Iobs
- "FC,PHIC": Fcalc
- "FOM,PHIW": figure of merit and weighted phase
- "SA,SIGSA":
 and error on
and error on - "FWT,PHWT": structure factors and phase terms for
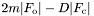
- "DELFWT,PHDELWT": structure factors and phase terms for
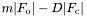
- debug
- none
- turn off debugging
- fileio
- output file I/O related debugging
- fit
- debugging output related to any minimization/fitting
- library
- all internal library information (library files/libpath/restraint internals) are output. Warning: can be VERY lengthy.
- memory
- all memory allocation/deallocation output.
- monomers
- all CIF library monomer information output. Warning: VERY lengthy.
- parser
- command line parser debugging.
- all
- turns on ALL debugging. Warning: extremely lengthy output.
- none
- calc
- redchi
- calculates the reduced chi based on several input models in a PDB file. Due to current restrictions, all models must be in one file, separated by "MODEL" instructions - the models themselves must increment starting at 1. See the PDB format documentation for further information.
- calculating the redchi value requires at least a multi-model input coordinate file, a fsigf and flag data from a reflection file (corresponding to observed data and a cross validation flag, see the MTZ symbols data types).
- due to the nature of parsing done during the redchi calculation, it is highly recommended to not combine this calculation with other calculation/execution commands.
- redchi
- refine
- mlf/realspace bfgs/bfgs2/cgpr/sd/anneal [ncycles]
- refine current parameters using a maximum likelihood reciprocal space target function/gradient (mlf) or real space function/gradient (realspace) for ncycles (default: 10) using the given refinement protocol (default: bfgs2). For simulated annealing, the ncycles argument is the number of tries per temperature cycle. For a further description of each minimizer protocol, see the descriptions for multidimensional minimizers and simulated annealing in the GSL manual. The recommended minimizer is BFGS or BFGS2, although if problems occur, the sd minimizer may provide faster results.
- refinement requires at least a model (such as a PDB file), a fsigf and flag data from a reflection file (corresponding to observed data and a cross validation flag, see the MTZ symbols data types).
- mlf/realspace bfgs/bfgs2/cgpr/sd/anneal [ncycles]
- graph "name1" ["name2"...]
- graph any of the currently read in/calculated data - the name must match any of the read in MTZ column labels, or use the default values for each data type (see the default types and MTZ symbols sections). Requires xmgrace support.
- set
- add_h on/off
- adds idealized hydrogen atoms to include in calculations (the atoms are not included in refinement, however)
- include_geom on/off
- include_xray on/off
- includes xray and geometry terms in refinement residuals/gradients
- restrain_phipsi on/off
- restrains phi and psi angles during refinement
- skip_bulksolvent on/off
- skips bulk solvent correction.
- write_maps on/off
- writes out and CCP4 format maps in addition to the MTZ file when "write mtz filename" is called, using the format "filename_2fo-fc.map" and "filename_fo-fc.map"
- writes out
- add_h on/off
- bins integer
- number of bins to carry out calculations with (default: 10)
- cvflag integer (default: 1)
- sets the integer value used to determine if a reflection is to be used for cross validation (i.e. Rfree).
- cell float float float float float float
- cell parameters: a b c alpha beta gamma
- if not entered, it will be read in from the first MTZ file read in.
- res float float
- resolution limits of data/calculations (in Å), can be in any order.
- if not entered, it will be determined from the resolution limits of the first MTZ file read in.
- sg "name or number"
- set the spacegroup for the data: must be listed using spaces between axes and cell setting (e.g. "P 21 21 21")
- if not entered, it will be read in from the first MTZ file read in.
- exit
- terminates parsing and exits
- fsigf/esige/isigi/fphi/phifom/flag "symbol,symbol..."
- set the MTZ column(s) to read in from the MTZ file(s). These can be listed in any order and at any time. The format depends somewhat on the type being read in. For example, the fsigf type only requires two parameters, the F column and the sigF column names, separated by a comma: fsigf "FO,SIGFO"
- the symbols can also include a dataset and crystal setting as well, such as:
- fsigf "native/xtal1/[FO,SIGFO]" would select crystal "native", dataset "xtal1" and columns "FO" and "SIGFO" from the mtz file
- fphi "*/*/[FC,PHIC]" would select any crystal and any dataset with a "FC" column (equivalent to simply using "fphi "FC,PHIC"")
- redchi will read in any and all data it finds from any of the MTZ input files with a matching column, and will output all of the data it reads in, in addition to any other data it computes (such as amplitudes and phases for difference maps, for example). For the purposes of using the read in data, redchi will use whatever type it needs that first matches the type required. So, for refinement, something like: "fsigf "FO,SIGFO" fsigf "FP,SIGFP"" would result in the program using FO and SIGFO for refinement, and only reading in the FP and SIGFP data.
- symbol translation/usage
- fsigf "col1,col2": amplitudes and errors
- esige "col1,col2": normalized amplitudes and errors
- isigi "col1,col2": intensities and errors
- fphi "col1,col2": amplitudes and phases
- phifom "col1,col2": phases and figure of merit
- flag "col": flagged data (e.g. freer)
- select "groupname" "selection" [or/and/xor "selection"...]
- set up a selection called "groupname" by selecting the atoms in "selection". Selections follow the CCP4 coordinate library syntax. Selections can be logical and/or/xor (exclusive or), so its easy to do:
select "group1" "A/*" or "B/*"
to select anything in chain A or B. - some default selections are provided, shown below:
- "default"
- selects all atoms - equivalent to "*" in CCP4 coordinate syntax
- "main"
- selects main chain atoms only - equivalent to "(GLY,ALA,VAL,PRO,SER,THR,LEU,ILE,CYS,ASP,GLU,ASN,GLN,ARG,LYS,MET,MSE,HIS,PHE,TYR,TRP)/N,CA,C,O,H,HA:*" in CCP4 coordinate syntax
- "side"
- selects side chain atoms only - equivalent to "(GLY,ALA,VAL,PRO,SER,THR,LEU,ILE,CYS,ASP,GLU,ASN,GLN,ARG,LYS,MET,MSE,HIS,PHE,TYR,TRP)/!N,CA,C,O,H,HA: *" in CCP4 coordinate syntax
- "default"
- the default selections can be used as selections themselves or as a groupname - e.g.:
- select "group1" "A/*" and "main"
- ("main" acts as selection in this case) - selects mainchain atoms in chain A
- xyz_refine individual "main"
- ("main" acts as a groupname in this case) - only refines mainchain atoms
- select "group1" "A/*" and "main"
- set up a selection called "groupname" by selecting the atoms in "selection". Selections follow the CCP4 coordinate library syntax. Selections can be logical and/or/xor (exclusive or), so its easy to do:
- NOTE: all anisotropic refinement currently does NOT work.
- xyz_refine
- off/group/individual ["groupname1"] ["groupname2"...]
- adp_refine
- off ["groupname1"] ["groupname2"...]
- auto ["groupname1"] ["groupname2"...]
- refines atoms anisotropically iff ANISOU parameters present (FIXME: currently broken)
- group/individual
- isotropic/anisotropic/tls ["groupname1"] ["groupname2"...]
- refines all atoms using a single tensor (group) or a tensor for each atom (individual) using the given tensor type (isotropic, anisotropic or TLS)
- isotropic/anisotropic/tls ["groupname1"] ["groupname2"...]
- by_residue integer isotropic/anisotropic/tls ["groupname1"] ["groupname2"...]
- refines a B-factor for each integer step of residues in the chain - e.g.:
- adp_refine by_residue 2 isotropic "default"
- refines a single isotropic B factor for 2 residue blocks in the chain
- adp_refine by_residue 2 isotropic "default"
- refines a B-factor for each integer step of residues in the chain - e.g.:
- occ_refine
- off/group/individual/auto ["groupname1"] ["groupname2"...]
- the "auto" refinement only refines occupancies in residues with alternate conformers or those in which the occupancy is *not* 0.0 or 1.0. Alternate conformers are constrainted to sum to 1.0 with an esd of 0.01.
- off/group/individual/auto ["groupname1"] ["groupname2"...]
- weight
- angle/bond/chiral/torsion/plane/bsim/anisobsphere float
- weights the named term based on the given number. (default for all weights: 1.0)
- df_xyz_b
- weights the xyz relative to the isotropic B gradient. Only applies when both xyz and isotropic Bs are refined simultaneously. (default: 0.01 - i.e. xyz is downweighted 100 fold relative to the B gradient)
- xray
- weights the xray probability AND gradient (default: 1.0)
- xray_df_xyz
- weights the x, y and z xray gradient terms (default: 1.0)
- xray_df_occ
- weights the occupancy xray gradient terms (default: 1.0)
- xray_df_iso
- weights the isotropic B factor xray gradient terms (default: 1.0)
- xray_df_aniso
- weights the anisotropic B factor xray gradient terms (default: 1.0)
- angle/bond/chiral/torsion/plane/bsim/anisobsphere float
- anneal_k float
- set the temperature multiplier in the kT divisor, larger values allow for larger model steps (default: 100.0)
- anneal_step float
- set the step size for simulated annealing (warning: this parameter affects the amount the model is varied in each trial, which can greatly alter the model if set to a large value) (default: 0.005)
- anneal_t0 float
- set the initial temperature for simulated annealing (default: 4000.0)
- anneal_dt float
- set the divisor in temperature per step (default: 1.5)
- ncs_restrain
- "ncsname" "groupname1" "groupname2" ["groupname3"...] [weight float]
- sets up a NCS restraint group called "ncsname" using the atom selections specified in the groupnames. Weights can optionally be provided (used in the same way as angle/bond/etc weights), otherwise the default weighting is 1.0. The weight reflects the expected rmsd between the NCS mates, so a value of 1.0 restraints the NCS mates to a rmsd of 1.0 Å.
- "ncsname" "groupname1" "groupname2" ["groupname3"...] [weight float]
#include <glib.h>
